Dementia refers to a set of symptoms that may include memory loss or difficulties in thinking, problem-solving or language. There is a common misconception that dementia is a normal part of ageing. Dementia only appears when our brains are impaired by neurodegenerative diseases. These diseases cause a loss of functions in brain cells. While these diseases mostly affect the older population, they can also affect younger individuals. The most known neurodegenerative diseases include Alzheimer’s Disease, Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. In Amyotrophic Lateral Sclerosis, mainly the systems that control muscles are impaired, which affects movement, breathing and swallowing.
Over the past 15 years, neurodegenerative diseases have become the leading cause of death worldwide, and the number of cases continues to rise in the UK. There are still no effective treatments for the neurodegenerative diseases that cause dementia, and many questions remain as to the root causes.
The UK Dementia Research Institute (UK DRI) is the single biggest investment the UK has ever made in neurodegenerative diseases, thanks to £290 million from founding funders the Medical Research Council (MRC), Alzheimer’s Society and Alzheimer’s Research UK. Revolutionary in scale and scope, and with collaboration at its core, the UK DRI brings together diverse expertise across seven centres nationally to accelerate the discovery, development and delivery of interventions that will help diagnose, treat and ultimately prevent dementia.
The UK DRI at King's was opened in August 2017 and is led by Centre Director Professor Jernej Ule. The mission of our centre is to understand the earliest molecular and pathophysiological events that initiate neurodegeneration and develop novel therapeutic strategies.
Our major disease interest is on Amyotrophic Lateral Sclerosis (ALS), Frontotemporal Dementia (FTD) and Alzheimer’s Disease (AD). Although distinct in the areas of the nervous system affected, these diseases do share common clinical pathological features. Our research looks at the similarities and differences between these diseases to find the keys to slow or halt loss of brain cells at the earliest stages.
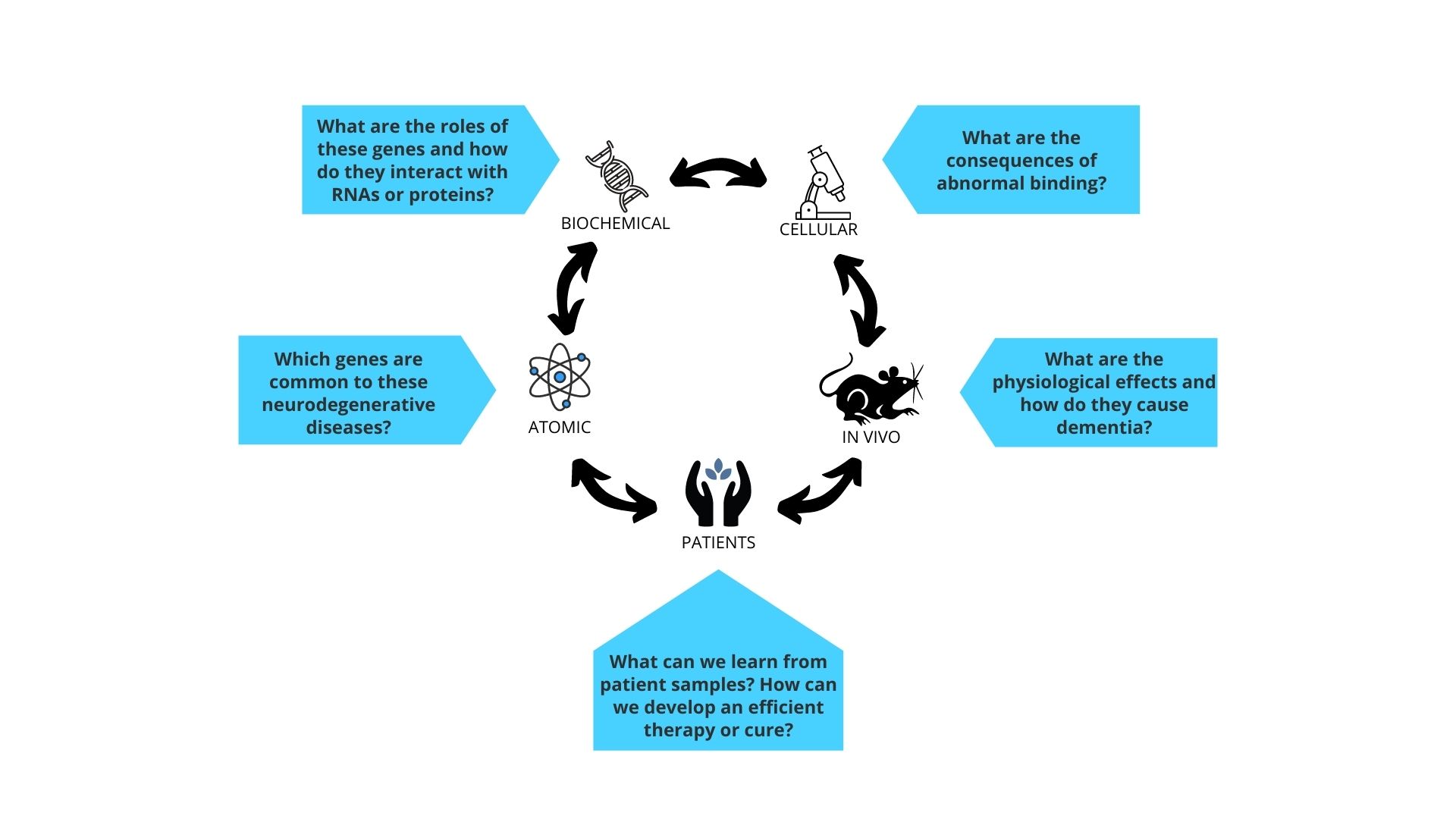
Our key questions:
- At the cellular level, which proteins in the human body are affected by neurodegenerative disease - what are the causes and consequences?
- How does disease affect the functions of brain cells such as the between neurons or the structural integrity of the neurons?
- What are the common pathways affected across different neurodegenerative diseases?
- What tools and technologies can we develop to improve the diagnosis and treatment of neurodegenerative disease?
- Can we use gene therapy to prevent or half the progression of neurodegenerative disease, and can we improve the way they are delivered??
The five UK DRI Group Leaders and their research focuses:
- Professor Jernej Ule (Centre Director) : Modulaton of protein-RNA complexes in in neurodegeneration
- Professor Chris Shaw : Gene discovery and gene therapy
- Dr Marc-David Ruepp: Altered RNA metabolism in neurodegeneration
- Dr Sarah Mizielinska: Nucleocytoplasmic transport dysfunction in FTD and ALS
- Dr Andrea Serio: Bioengineered platforms to model neurodegeneration in single cells and circuits”
Learn more:
 Members of our centre at the UK DRI Connectome 2019.
Members of our centre at the UK DRI Connectome 2019.
'Between the signals' podcast
UK Dementia Research Institute at King’s College London has launched a new podcast, ‘Between the signals’ which is aimed at bringing greater understanding about neurodegenerative conditions, and the groundbreaking research going into treating them to a public audience.
In the first episode 'What is motor neuron disease?', Dr Asma Bashir (King’s College London) and Dr Sarah Marzi speak to Dr Sarah Mizielinska to understand more about the condition including common symptoms, diagnosis and current treatments. The episode coincides with Global MND Awareness Day which falls on 21 June.
You can now listen and watch episode one on YouTube or listen via the following podcast platforms.